High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation …
Köhler, Tim et al. (2012),
Applied and Environmental Microbiology,
vol. 78,
4691-4701
Köhler, Tim, Dietrich, Carsten, Scheffrahn, Rudolf H., Brune, Andreas (2012),
Applied and Environmental Microbiology,
vol.
78,
4691-4701
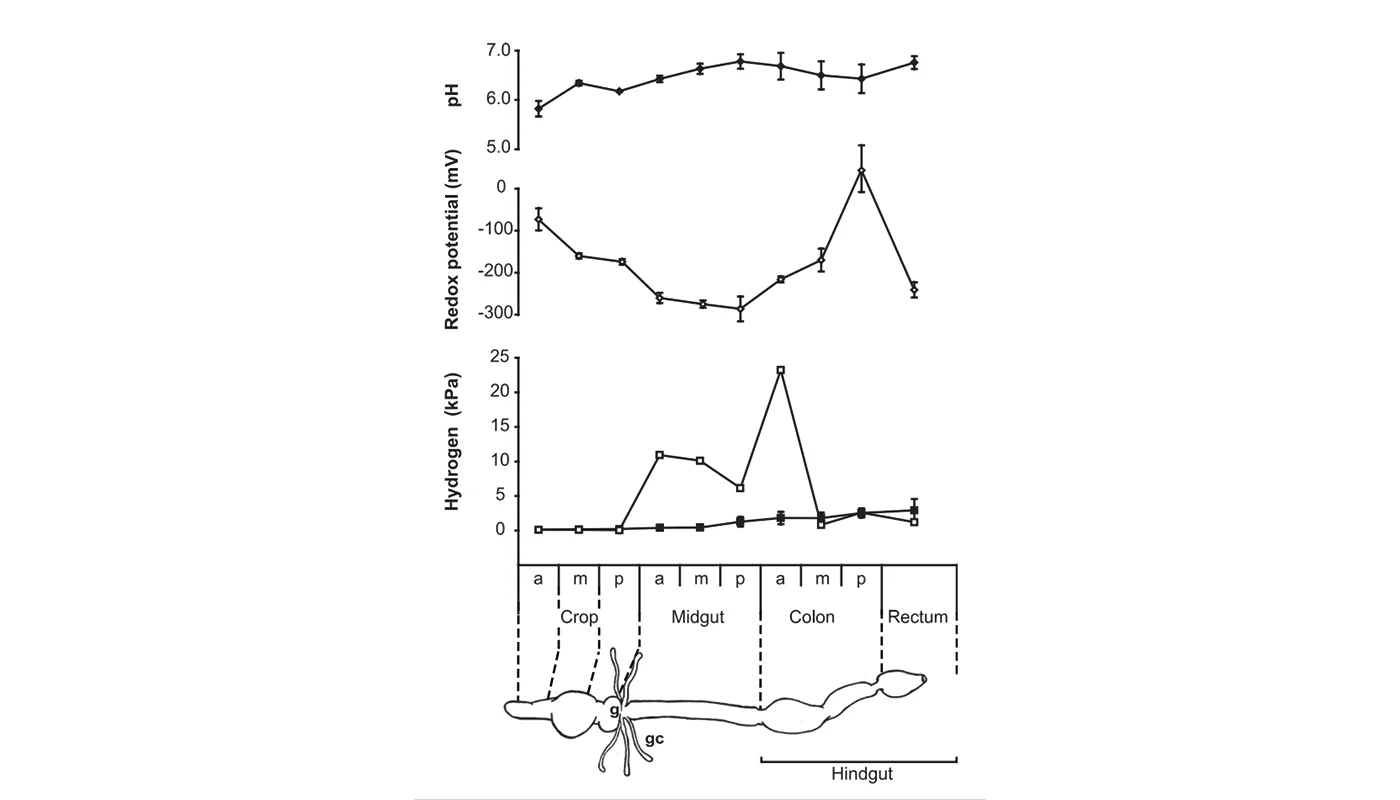
Higher termites are characterized by a purely prokaryotic gut microbiota and an increased compartmentation of their intestinal tract. In soil-feeding species, each gut compartment has different physicochemical conditions and is colonized by a specific microbial community. Although considerable information has accumulated also for wood-feeding species of the genus Nasutitermes, including cellulase activities and metagenomic data, a comprehensive study linking physicochemical gut conditions with the structure of the microbial communities in the different gut compartments is lacking. In this study, we measured high-resolution profiles of H2, O2, pH, and redox potential in the gut of Nasutitermes corniger termites, determined the fermentation products accumulating in the individual gut compartments, and analyzed the bacterial communities in detail by pyrotag sequencing of the V3-V4 region of the 16S rRNA genes. The dilated hindgut paunch (P3 compartment) was the only anoxic gut region, showed the highest density of bacteria, and accumulated H2 to high partial pressures (up to 12 kPa). Molecular hydrogen is apparently produced by a dense community of Spirochaetes and Fibrobacteres, which also dominate the gut of other Nasutitermes species. All other compartments, such as the alkaline P1 compartment (average pH, 10.0), showed high redox potentials and comprised small but distinct populations characteristic for each gut region. In the crop and the posterior hindgut compartments, the community was even more diverse than in the paunch. Similarities in the communities of the posterior hindgut and crop suggested that proctodeal trophallaxis or coprophagy also occurs in higher termites. The large sampling depths of pyrotag sequencing in combination with the determination of important physicochemical parameters allow cautious conclusions concerning the functions of particular bacterial lineages in the respective gut sections to be drawn. © 2012, American Society for Microbiology.
10.1128/AEM.00683-12
Phylogenetic diversity, localization, and cell morphologies of members of the candidate phylum TG3 and a …
Brune, Andreas. et al. (2006),
Applied and Environmental Microbiology,
vol. 72,
6780-6788
Brune, Andreas., Emerson, D., Breznak, J. A., Hongoh, Yuichi, Deevong, Pinsurang, Hattori, Satoshi, Inoue, Tetsushi, Noda, Satoko, Noparatnaraporn, Napavarn, Kudo, Toshiaki, Ohkuma, Moriya (2006),
Applied and Environmental Microbiology,
vol.
72,
6780-6788
Recently we discovered two novel, deeply branching lineages in the domain Bacteria from termite guts by PCR-based analyses of 16S rRNA (Y. Hongoh, P. Deevong, T. Inoue, S. Moriya, S. Trakulnaleamsai, M. Ohkuma, C. Vongkaluang, N. Noparatnaraporn, and T. Kudo, Appl. Environ. Microbiol. 71:6590-6599,2005). Here, we report on the specific detection of these bacteria, the candidate phylum TG3 (Termite Group 3) and a subphylum in the phylum Fibrobacteres, by fluorescence in situ hybridization in the guts of the wood-feeding termites Microcerotermes sp. and Nasutitermes takasagoensis. Both bacterial groups were detected almost exclusively from the luminal fluid of the dilated portion in the hindgut. Each accounted for approximately 10% of the total prokaryotic cells, constituting the second-most dominant groups in the whole-gut microbiota. The detected cells of both groups were in undulate or vibroid forms and apparently resembled small spirochetes. The cell sizes were 0.2 to 0.4 by 1.3 to 6.0 μm and 0.2 to 0.3 by 1.3 to 4.9 μm in the TG3 and Fibrobacteres, respectively. Using PCR screenings with specific primers, we found that both groups are distributed among various termites. The obtained clones formed monophyletic clusters that were delineated by the host genus rather than by the geographic distance, implying a robust association between these bacteria and host termites. TG3 clones were also obtained from a cockroach gut, lake sediment, rice paddy soil, and deep-sea sediments. Our results suggest that the TG3 and Fibrobacteres bacteria are autochthonous gut symbionts of various termites and that the TG3 members are also widely distributed among various other environments. Copyright © 2006, American Society for Microbiology. All Rights Reserved.
10.1128/AEM.00891-06
The bacterial community in the gut of the cockroach Shelfordella lateralis reflects the close evolutionar…
Schauer, Christine et al. (2012),
Applied and Environmental Microbiology,
vol. 78,
2758-2767
Schauer, Christine, Thompson, Claire L., Brune, Andreas (2012),
Applied and Environmental Microbiology,
vol.
78,
2758-2767
Termites and cockroaches are closely related, with molecular phylogenetic analyses even placing termites within the radiation of cockroaches. The intestinal tract of wood-feeding termites harbors a remarkably diverse microbial community that is essential for the digestion of lignocellulose. However, surprisingly little is known about the gut microbiota of their closest relatives, the omnivorous cockroaches. Here, we present a combined characterization of physiological parameters, metabolic activities, and bacterial microbiota in the gut of Shelfordella lateralis, a representative of the cockroach family Blattidae, the sister group of termites. We compared the bacterial communities within each gut compartment using terminal-restriction fragment length polymorphism (T-RFLP) analysis and made a 16S rRNA gene clone library of the microbiota in the colon-the dilated part of the hindgut with the highest density and diversity of bacteria. The colonic community was dominated by members of the Bacteroidetes, Firmicutes (mainly Clostridia), and some Deltaproteobacteria. Spirochaetes and Fibrobacteres, which are abundant members of termite gut communities, were conspicuously absent. Nevertheless, detailed phylogenetic analysis revealed that many of the clones from the cockroach colon clustered with sequences previously obtained from the termite gut, which indicated that the composition of the bacterial community reflects at least in part the phylogeny of the host. © 2012, American Society for Microbiology.
10.1128/AEM.07788-11
Gut anatomical properties and microbial functional assembly promote lignocellulose deconstruction and col…
Ceja-Navarro, J.A. et al. (2019),
Nature Microbiology,
vol. 4,
864-875
Ceja-Navarro, J.A., Karaoz, U., Bill, M., Hao, Z., White III, R.A., Arellano, A., Ramanculova, L., Filley, T., Berry, T., Conrad, M., Blackwell, M., Nicora, C., Kim, Y.M., Reardon, P., Lipton, M., Adkins, J.N., Pett-Ridge, J., Brodie, E.L., Ceja-Navarro, Javier A., Karaoz, Ulas, Bill, Markus, Hao, Zhao, White, Richard A., Arellano, Abelardo, Ramanculova, Leila, Filley, Timothy R., Berry, Timothy D., Conrad, Mark E., Blackwell, Meredith, Nicora, Carrie D., Kim, Young Mo, Reardon, Patrick N., Lipton, Mary S., Adkins, Joshua N., Pett-Ridge, Jennifer, Brodie, Eoin L. (2019),
Nature Microbiology,
vol.
4,
864-875
Beneficial microbial associations enhance the fitness of most living organisms, and wood-feeding insects offer some of the most striking examples of this. Odontotaenius disjunctus is a wood-feeding beetle that possesses a digestive tract with four main compartments, each of which contains well-differentiated microbial populations, suggesting that anatomical properties and separation of these compartments may enhance energy extraction from woody biomass. Here, using integrated chemical analyses, we demonstrate that lignocellulose deconstruction and fermentation occur sequentially across compartments, and that selection for microbial groups and their metabolic pathways is facilitated by gut anatomical features. Metaproteogenomics showed that higher oxygen concentration in the midgut drives lignocellulose depolymerization, while a thicker gut wall in the anterior hindgut reduces oxygen diffusion and favours hydrogen accumulation, facilitating fermentation, homoacetogenesis and nitrogen fixation. We demonstrate that depolymerization continues in the posterior hindgut, and that the beetle excretes an energy- and nutrient-rich product on which its offspring subsist and develop. Our results show that the establishment of beneficial microbial partners within a host requires both the acquisition of the microorganisms and the formation of specific habitats within the host to promote key microbial metabolic functions. Together, gut anatomical properties and microbial functional assembly enable lignocellulose deconstruction and colony subsistence on an extremely nutrient-poor diet.
10.1038/s41564-019-0384-y
Compartmentalization of bacterial and fungal microbiomes in the gut of adult honeybees
Callegari, Matteo et al. (2021),
npj Biofilms and Microbiomes,
vol. 7,
-
Callegari, Matteo, Crotti, Elena, Fusi, Marco, Marasco, Ramona, Gonella, Elena, De Noni, Ivano, Romano, Diego, Borin, Sara, Tsiamis, George, Cherif, Ameur, Alma, Alberto, Daffonchio, Daniele (2021),
npj Biofilms and Microbiomes,
vol.
7,
-
The core gut microbiome of adult honeybee comprises a set of recurring bacterial phylotypes, accompanied by lineage-specific, variable, and less abundant environmental bacterial phylotypes. Several mutual interactions and functional services to the host, including the support provided for growth, hormonal signaling, and behavior, are attributed to the core and lineage-specific taxa. By contrast, the diversity and distribution of the minor environmental phylotypes and fungal members in the gut remain overlooked. In the present study, we hypothesized that the microbial components of forager honeybees (i.e., core bacteria, minor environmental phylotypes, and fungal members) are compartmentalized along the gut portions. The diversity and distribution of such three microbial components were investigated in the context of the physico-chemical conditions of different gut compartments. We observed that changes in the distribution and abundance of microbial components in the gut are consistently compartment-specific for all the three microbial components, indicating that the ecological and physiological interactions among the host and microbiome vary with changing physico-chemical and metabolic conditions of the gut.
10.1038/s41522-021-00212-9